From Rosetta@home to Folding@home
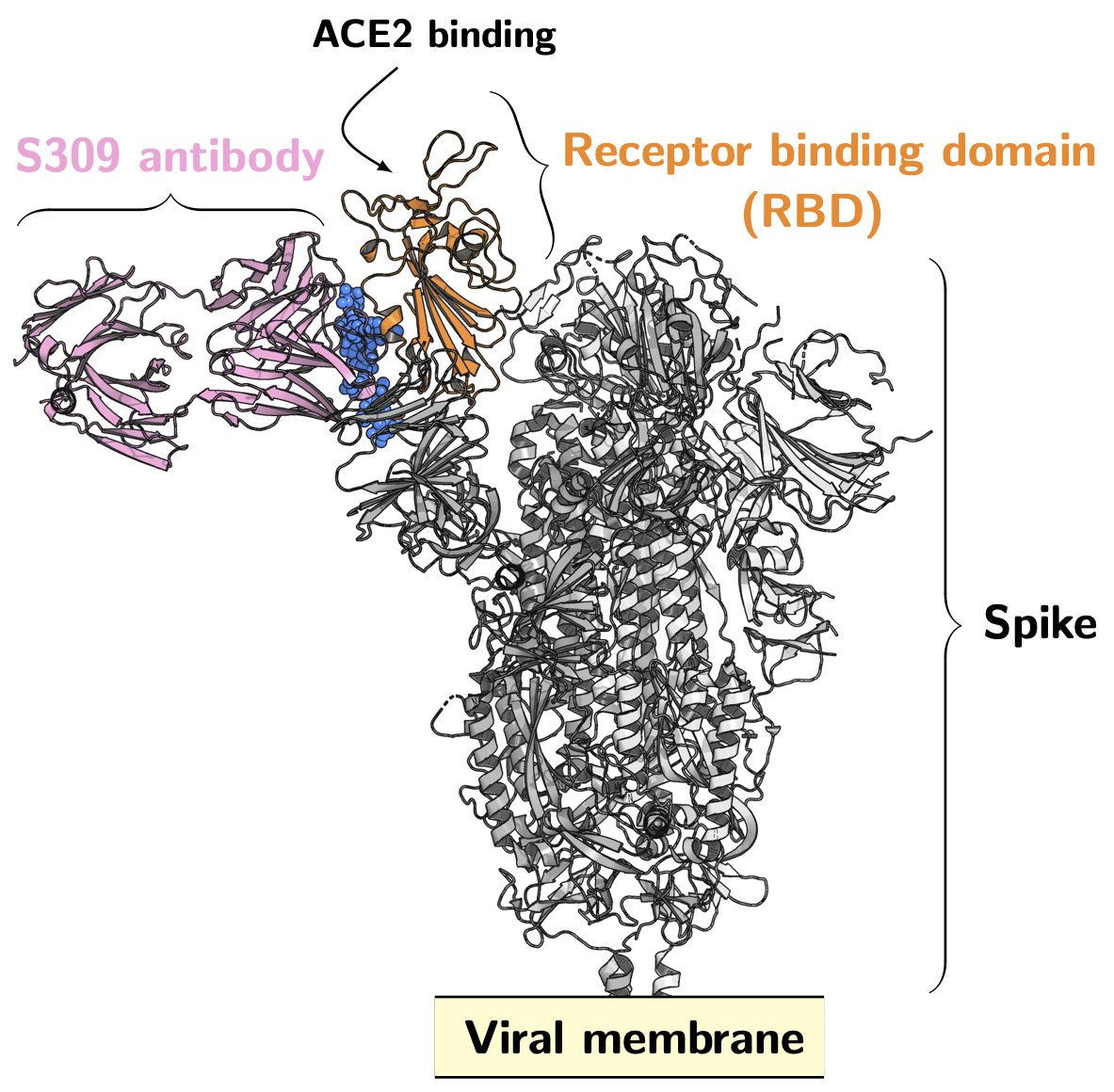
Let’s start with the main principle. Rosetta@home is only predicting the molecular structure of synthetic drugs capable of Binding Covid-19 outer spike-in protein, which can be imagined as similar to the photographic technique in photography. Rosetta can statically predict the characteristics of a particular protein, but it cannot predict how that protein molecule will function under the evolution of time. Because there will be potential differences between molecules that generate electromagnetic forces, and all the constituent elements within the molecule will interact with each other to create motion over time, when we design a synthetic drug in Rosetta that can bind the SARS-COV-2 virus, we need to enter the testing phase to see if the drug can bind to the viral protein stably, because the predicted structure is static, we cannot know whether the protein structure can bind well to the viral protein under the interaction motion, here Folding@home comes in handy, it is similar to shooting a motion picture in photography, we can get the static protein structure chain from Rosetta@home, and then simulate in Folding@home whether that protein binds well to the viral protein over time to produce resistance to the virus.
Join the project

I will shut down Rosetta@home, which has been running before, and now Rosetta@home has published a paper predicting the artificial protein structure that can bind SARS-COV-2, so all I need to do now is to do a lot of molecular dynamics simulations, and Folding@home already supports ARM64 architecture, I will migrate all the computing power I run to Folding@home, expecting the number of infections worldwide to come down, enabling the economy to recover.